












































納品物:
4 μg の凍結乾燥プラスミド(1 μg、低コピープラスミド用)*
電子ベクターマップ
*プラスミドコピーをご確認ください
品質管理:
お客様のカスタムインサートを含むシーケンスクロマトグラム
品質保証証明書

GenScriptは、CRISPR技術のライセンスを、Broad Institute of MIT and Harvardより取得しています。当社の提供品には、CRISPRのパイオニアであるFeng Zhang研究室によって開発された、最新のCRISPRプラスミドおよびデータベースがあります。Broad Institute検証済のプラスミドはCRISPR/Cas9発現用の十分に試験されたプラットフォームで、またRNAベースのプラットフォームにおける不安定性の問題を回避します。

GenScript CRISPRプラスミドコレクション
GenScriptは、Broad Instituteに事前検証されたガイドRNA(gRNA)配列を含む、20,000点以上のlentiCRISPRv2プラスミドを維持しています。プラスミドは遺伝子名、シンボルあるいはIDで、当社gRNAデータベースから検索可能です。
| Product | Vector | Selection | |
|---|---|---|---|
| GenCRISPR™ Plasmid Collection | Lentiviral | Amp, Puro | Broad gRNA Database |
Enhanced CRISPR/SpCas9New!
SpCas9(K848A/K1003A/R1060A)とも呼ばれる、特異性がEnhanced CRISPR/SpCas9 (eSpCas9)は、Broad InstituteのFeng Zhang研究室の研究者らにより、ターゲットへの特異性が向上する構造的改変がされています(Slaymaker et al. 2016)。eSpCas9は、しっかりとしたオンターゲットゲノム編集効率を維持しながらも、オフターゲット効果を10倍以上低減できます。
詳細情報 »
Cas9 genome editing is dependent on the separation of DNA double strands. Mismatches between sgRNA and untargeted DNA sequences can cause unspecific binding and cleavage. To improve genome editing specificity, SpCas9 with mutations K848A, K1003A, and R1060A was developed. Neutralization of these positively charged residues within the non-target strand groove of SpCas9 weakened non-target binding and encouraged on-target binding which requires more stringent Watson-Crick base pairing.
| Product | Vector | Selection | |
|---|---|---|---|
| eSpCas9 Plasmids | Plasmid Lentiviral |
Puro or GFP | お問合せ |
SpCas9プラスミド
Cas9エンドヌクレアーゼは、遺伝子編集における研究スタンダードです。シングルガイドRNA(sgRNA)配列と組み合わせた際、これらの酵素はゲノム中に部位特異的な二本鎖切断(DSBs)を生じさせます。
詳細情報 »
SpCas9 also contains on-target affinity for NGA sequences.
| Product | Vector | Selection | |
|---|---|---|---|
| SpCas9 Plasmids | Plasmid Lentiviral AAV |
Amp Amp, Puro Amp, Neo Amp, GFP |
お問合せ |
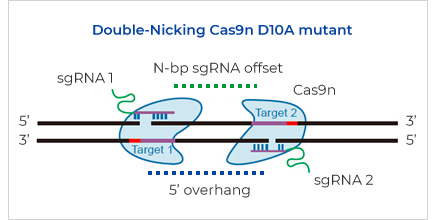
SpCas9ニッカーゼプラスミド
SpCas9ニッカーゼ(Cas9n D10A)は、DSBsとは対照的に、エンドヌクレアーゼにより一本鎖のニックを生じることができる変異を含んでいます。二つの反対側に向いたgRNA配列をSpCas9ニッカーゼ配列と組み合わせることは、不要なインデルの形成を防ぐ、効率的な遺伝子編集の方法です。
| Product | Vector | Selection | |
|---|---|---|---|
| SpCas9 Nickase Plasmids | Plasmid Lentiviral |
Amp Amp, Puro Amp, GFP |
お問合せ |
SaCas9プラスミド
黄色ブドウ球菌Cas9オルソログ(SaCas9)は、アデノ随伴ウイルス(AAV)のアプリケーションにふさわしいエンドヌクレアーゼです。SaCas9はSpCas9よりも約1 kb短く、またAAVのパッケージングにおける制限などに、さらなる柔軟性を提供します。AAVベクターのより低い免疫原性により、SaCas9はin vivoでの編集アプリケーションや治療に最適です。
詳細情報 »
The optimal on-target SaCas9 PAM sequence is NNGRRT.
SaCas9 also contains significant on-target affinity for NNGRRN.
| Product | Vector | Selection | |
|---|---|---|---|
| SaCas9 Plasmids | AVV | Amp | お問合せ |
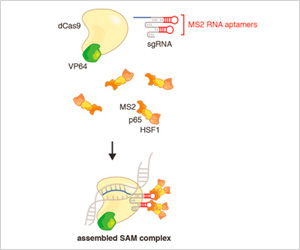
転写活性化(SAM)プラスミド
CRISPR/Cas9相乗的活性化メディエーター(SAM)システムは、ダウンストリームターゲットの転写活性化を可能にするよう改変されています。SAMシステムは、触媒的に活性のないCas9(dCas9)複合体上に組み立てられた3つの異なる活性化因子:VP64、P65、およびHSF1を利用して、転写を促進します。
詳細情報 »
The SAM complex is comprised of three components: a gRNA incorporating two MS2 RNA aptamers, a catalytically inactive dCas9-VP64 fusion protein, and a MS2-P65-HSF1 activator fusion protein.
The SAM system is capable of activation of both coding and non-coding genetic elements.
To search for Broad Institute pre-validated SAM gRNA sequences, visit our guide RNA Database.
| Product | Vector | Selection | |
|---|---|---|---|
| SAM gRNA Plasmids | Plasmid Lentiviral |
Amp Amp, Zeo |
お問合せ |
| SAM dCas9-VP64 Plasmids | Lentiviral | Amp, Blast Amp, GFP |
お問合せ |
| SAM MS2-P65-HSF1 Plasmids | Lentiviral | Amp, GFP Amp, Hygro |
お問合せ |
Broad Instituteプラスミドコレクション
Broad Instituteプラスミドは、HarvardとMITのBroad Instituteによって作られました。これらのプラスミドは、カスタマイズされたsgRNA配列の代わりに17 bp-1.8 kbの発現可能なリンカーを含んでおり、お客様の研究室で改変することができます
